
和mRNA和蛋白一样,细胞内不同miRNAs的表达水平区别很大。总体而言,成熟的miRNAs半衰期以星期计[10],所以其表达量主要由miRNAs产生的速率,即图1的RNA转录和加工步骤来决定。许多miRNAs基因的转录因子已被报道[11,12]。另外,某些RNA结合蛋白,如Lin28,可与特定的pri-miRNAs或pre-miRNAs结合进而影响RNA的加工和miRNAs的成熟[12]。这些研究成果可以解释miRNAs表达的细胞、组织特异性,或同一miRNAs在细胞不同状况、不同处理条件下的表达差异。但是,它们不能解释为什么基因组所有miRNAs的表达量会千差万别。此类问题是功能基因组学和系统生物学最重要的问题之一。例如,研究最久、颇具争议的,mRNA水平与相应蛋白水平之间的关系,即蛋白质表达量到底由什么因素决定?一些研究发现酵母和哺乳动物细胞中全基因组范围内的mRNA丰度和蛋白丰度的相关性约为0.6[13,14],说明蛋白表达量主要在翻译的层次上调控。而近来的研究则认为mRNA丰度和蛋白丰度的相关性可高达0.9[15-17],表明转录对基因表达的贡献最大。
虽然转录活性通常被认为在决定miRNAs的整体表达模式中起到重要作用,但直接证据是缺乏的。鉴定个体miRNAs的特异性转录因子本质上并不能解决为什么特定的miRNAs比其他miRNAs的丰度更高或低的问题。
功能基因组学和系统生物学为生物学的研究开阔了新途径,在基因组的水平上解析miRNAs表达的调控。目前已有研究者进行了相关工作[8,18,19],采取了不同以往的研究策略和思路,得到在以前无法获得或不可想象的结论。近年来,曾严等[8]分析了在基因组水平上转录活性和miRNAs表达量之间的关系,发现在人的K562细胞系中相关系数仅为0.3,而在另一细胞系,HeLa,没有显著的相关性。该研究结果始料未及,因为之前并没有这方面的报道,前人一直只是假设转录为miRNAs表达的决定性因素。
本试验极大地巩固和发展现有成果,利用人的细胞系为研究对象,进一步研究清楚基因组水平上转录活性和miRNAs表达量之间的关系,进一步探讨转录对miRNAs差异性表达的影响,以揭示调控机制的保守性及重要性,该成果可以应用到其它非编码RNA、生物系统和领域,为今后的工作指出新的方向,科学意义广泛。
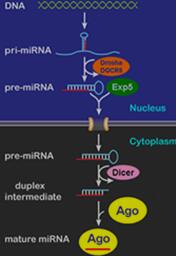
图1 典型动物miRNAs的形成途径[8]
1 资料与方法
1.1 实验数据的下载、分析平台
1.1.1数据下载网页平台
e!ensembl网站下载“人类基因组信息数据集”,版本:GRCh38,下载网址为:http://asia.ensembl.org/biomart/martview/27191896953a3ac779448a3f113a5a8b);
基因组miRNAs信息(“人类miRNAs基因组数据集”)参考miRBase[9];
于ENCODE网站(网址为https://www.encodeproject.org/)以及NCBI的Gene Expression Omnibus网站(网址为https://www.ncbi.nlm.nih.gov/gds)下载人类细胞系各自对应的mRNA-seq和microRNA-seq数据文件、ChIP-seq和microRNA-seq数据文件,若无microRNA-seq,则用small RNA-seq代替。
1.1.2数据分析平台及软件
Excel数据分析软件
PAST数据分析工具
Galaxy网页数据分析平台(网址为https://usegalaxy.org/)
1.2 实验方法
1.2.1查找基因组数据集
e!ensembl网站下载所需的“人类基因组信息数据集”文件,所需的内容包含以下:基因所在染色体上的位置,即染色体数、基因起始与终止位点、基因ID、基因名。基因组miRNAs信息参考miRBase[9],部分pri-miRNAs已有实验证据,则可用相应信息代替[20,21]。
1.2.2 查找人类各细胞系对应的miRNA-seq、mRNA-seq和ChIP-seq的数据集 以人的细胞系为模型分析miRNAs表达量与转录的相关性(2):http://www.751com.cn/shengwu/lunwen_21893.html