- 上一篇:水下目标激光探测的Monte Carlo模拟研究+MATLAB程序
- 下一篇:ANSYS基于电磁加热熔胶系统研究
本论文在阐述了与石墨型C4N3相关的半金属性、无金属磁性、泛函密度分析等理论以及模拟软件VASP后,通过VASP软件计算,对在应变作用下C4N3的电学特性和磁学特性进行了计算研究。
12 C4N3的几何结构
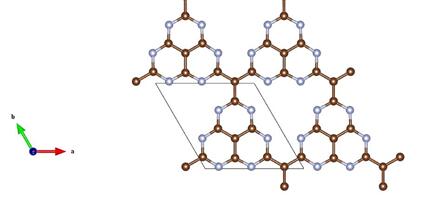
在C4N3有潜力应用于燃料电池,光催化,和制氢和g- C4N3显示器ferromagnetism。因此研究C4N3的物性随各种条件的变化而变化是很有意义的。要研C4N3究物性变化,就要研究掌握其几何结构。然而,一个系统的稳定性研究C-N的电子结构、光学性质和磁性基于材料仍然缺乏。先前的研究发现g- C4N3有几个结构同分异构体,可以分类成两个家庭根据他们不同的杂环块:单码道三嗪或三环heptazine [13],展示不同的电子和磁特性因此有不同的潜在应用。我们研究的材料正常情况下的几何结构如图。
深色的表示C原子,浅色的表示N原子
13 C4N3的研究现状
本文的研究背景
综上所述,首先,g- C4N3是一种备受关注的材料,自旋电子学则是本世纪极具潜力的新兴学科,因此研究g- C4N3的电磁特性随应变的变化一直是热点工作之一。目前,已经从理论上预测了g- C4N3具有无金属磁性和本金属性。
近来,由于其在自旋电子学的应用潜力,过渡金属无磁性和半金属性一直是的研究活动的重要主题。在这里,我们第一次通过泛函密度理论表明,最近实验实现石墨碳氮化(g - C4N3)显示一个铁磁基态。此外,这种新型材料,预计拥有内在半金属性至今没有报告。我们的研究结果突出表明了这种材料对现实不含金属的自旋电子学的应用的新希望。
最后,根据第一原理仿真中,最近实验证实了g- C4N3材料[35]的铁磁基态和内在的半金属性。以实验的方法合成不含金属的半金属性物质可能难以控制,因为必须要一个强大的外部电流或仔细选择性掺杂。尽管实验合成的纯g- C4N3可能仍然是一个挑战。作为不含金属的磁铁由于小自旋轨道耦合可能提供大自旋弛豫时间,结果凸显了新材料朝向现实无金属自旋电子学应用的实验验证研究的新希望。
15本文的主要工作
针对上述问题,本文选取了实验合成且在通常环境下能够稳定存在的g -C4N3做为研究对象,目的在于尽量避免仅仅用理论设计预测的材料能否被实验成功合成这一问题。采用第一性原理计算方法,从理论上系统研究应变对g- C4N3的电磁特性等物理特性的影响,从而掌握其变化规律进而指导g- C4N3材料的应用,本文的主要工作分为以下几个部分:
1、了解密度泛函理论
2、了解电子自旋学
3、收集有关g- C4N3材料的相关信息
4、了解VASP相关知识
2 理论基础和计算软件
21 密度泛函理论
采用近似和简化的方法处理固体中原子核和电子的数量很庞大(一般数量级都在1029 )的微观物理问题 。电子和原子核的多粒子系统分开需要通过波恩•奥本海默绝热近似;多电子问题简化为单电子问题要通过Hartree-Fock 自洽场方法。密度泛函理论可以更精确的说明这一问题。[23-24]
1Thomas-Fermi的模型
密度泛函理论产生可以上溯到1927年Thomas和Fermi提出的Fermi‐Thomas模型。他们为了计算原子的能量,将一个原子的动能表达成原子核-电子和电子-电子相互作用和电子密度的泛函。当初Fermi和Thomas用统计方法原理将院子动能近似地表示原子中电子分布,并提出假设:电子运动的751文相空间中,每个体积元中都均匀分布着两种电子,有电子和原子核分布确定一个有效势场。这样的假设,就可以得到Fermi-Thomas公式。这个假设模型是很重要的第一步,但由于没有考虑原子交换能(Hartree‐Fock 理论),它的精度受到了限制。1928年Dirac在该模型基础上增加一个交换能泛函项。然而,在很多数应用中Thomas‐Fermi‐Dirac理论表现得非常不精确,然后是对电子关联作用的完全忽略,以及交换能中的误差,导致动能的误差很大。